X射线光电子能谱XPS
仪器型号: Thermo Kalpha;Thermo ESCALAB 250XI; Axis Ultra DLD Kratos AXIS SUPRA; PHI-5000versaprobeIII等
1、样品状态:可为粉末、块状、薄膜样品;
2、粉末样品:20-30mg;
3、块状、薄膜样品:块体/薄膜样品尺寸小于5*5*3mm;
4、液体样品:最好自己掌握浓度制样,如果需要我们制样,需要保证一定浓度,否则测试结果不好,自行负责!
5、特殊需求:样品含S、F、I、Br等元素单质,如果容易挥发则不能测试,具体联系工程师;
6、样品包装要求:样品制备好以后,尽可能进行真空密封,减少样品吸附空气中的污染物,对于某些元素的测试,会有影响。
7、XPS测试需要样品用导电胶固定在样品台上,所以测过的粉末样品没法回收,块体样品回收可能也受到污染或者破坏,建议尽量不回收样品。
1、测试检测结果给出的是VGD格式和EXCEL格式测试结果。EXCEl格式文件用ORIGIN软件作图;VGD格式文件可以用Avantage分析软件打开。
-20211118184437.jpg)
2、全谱分析和窄区扫描
-20211118184544.jpg)
1、对于C窄谱要不要测?C窄谱对数据直接的影响是什么?
答:可以不测,但是都用于校正,也可以用O的531.1eV,或者金的零价标准峰进行校正。
2、有些客户不用测C,但是在peak table 里加了C元素。他能否直接C的百分扣除,直接去重新计算其他元素的百分比?还是需要复测或者用测试老师这里的机器去导出数据,重新处理?
答:可以直接去除的,不需要复测,或者重新导出也可以。
3、数据中C、N、O元素含量比实际样品的高,或没有C、N、O但是测出来了?
(1)样品吸附了空气中的污染物导致,在数据处理时,把非样品所含有的污染物的峰先分出来,剔除掉 ,再重新计算含量;
(2)如果对这个要求较严,建议后期样品制备后抽真空密封保存,测试检测时选择手套箱制样并测试;如果是非粉体样品,可选择刻蚀后测试。
4、样品不含有这个元素,为什么全谱测出这个元素了(除CNO)?
(1)样品理论上应该不含有,但是样品在处理过程中是有引入该元素的成分,在后续的处理过程没有完全处理干净;
(2)待测试样品是在某种基底上测试的,但待测试样品量较少,测试到了基底;
(3)被同一批次的其他样品,或者有易挥发组分(如S、F),或者进行刻蚀后,污染了该样品表面。
5、某元素含量很高大概 5%,测出来信噪比很差,而有的元素含量很低0.5%,测出来信噪比反而很好?
(1)不同元素的主峰的灵敏度因子和检测限不一样;
(2)样品分布不均匀;
(3)该元素不分布在样品表面,即不在XPS所测微区范围内。
6、所测窄谱元素,含量比为什么跟预期不符?
(1)样品表面被污染;
(2)样品分布不均匀;
(3)该元素不分布在样品表面,即不在XPS所测微区范围内;
(4)XPS是半定量分析,和元素的实际含量会有出入。
PS:XPS是一种典型的表面分析手段,用于定性及半定量分析,测试得到的仅是样品表面几百甚至几十um大小,几个nm深度的样品信息,不代表样品整体性质。
7、样品中,某元素窄谱曲线不光滑,峰刺较多,或没有测出该元素?
(1)元素含量比较低;
(2)样品分布不均匀,所测试的光斑范围内,该元素含量较少;
(3)该元素不分布在样品表面,即不在XPS所测微区范围内。
8、分峰后,某元素结合能位置不对?
(1)确认下数据是否校正(我们给的数据,一般是没有校正的数据);
(2)分析下化学环境对该元素峰本身造成的影响,某些化学环境会导致峰有正常的偏移;
(3)确认下分峰是否正确。
9、元素的结合能测试范围和之前的不同,无法对比?
答:如果该元素的峰是完整的,可以自行截取至相同结合能范围进行对比,如果不影响分析也可以不进行截取。
10、分峰后,对应价态的峰没有?
(1)需要分峰后进行分析,并需确认分峰是否正确;
(2)全谱的能量很高,如果测不出来,可能是污染碳很高,或者测试深度范围内的含量太少(整体该价 态比较高,但是表面很少),一般是后者原因居多。
11. 样品精细谱扫出谱峰?为什么全谱里没有呢?
全谱主要是用来定性分析的,设置参数的步长比较大,含量低的在全谱里扫不出谱峰。但是精细谱扫出谱峰就表示有该元素。
12. 每种元素的检测限一样么?
不一样。每种元素的主峰的灵敏度因子都不一样。
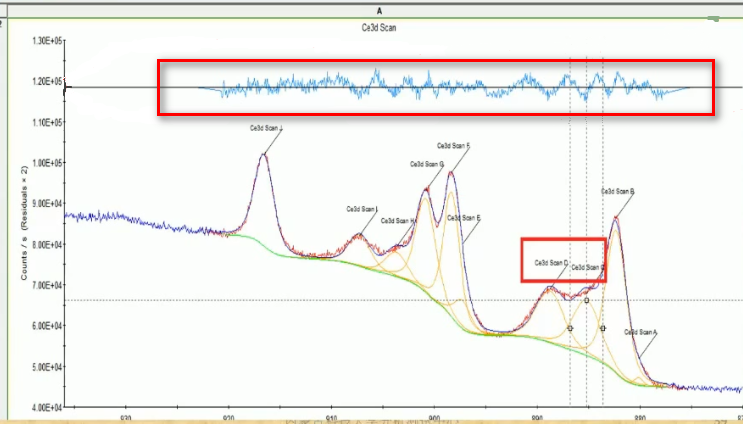
13. 怎么判断拟合是好是坏,是拟合了两个峰算好还是拟合了三个峰算好?
看波动大小,越小越好;还要看对应的物理意义。波动如下图所示。具体拟合几个峰,要参考样品本身的情况,以及拟合的贴合度,没有严格的界定哪个更好。